



摘要
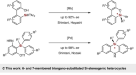
C−Si键的裂解是硅基各种转化的关键步骤之一。然而,未应变Si−C(sp3.由过渡金属催化的化学键仍处于初级阶段。它们通常包括插入一个M−C(sp2(C - Si键)和相应的分子内C(sp2) -Si耦合,专门生产硅井。在此我们报道了pd催化的硅螺环化反应,其中Si - C(sp3.)键通过插入M−C(sp3.)种,然后形成新的C(sp3.) -Si键,允许构造各种螺旋硅环。DFT计算强烈支持的这种反应模式可能为Si−C(sp3.)键解和硅环合成。
简介
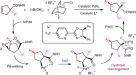
开发能够催化C-Si键裂解的方法是过渡金属催化的长期目标1,2,3.,4,5因为它对许多学科中使用的有机硅化合物的反合成分析和后续合成有重大影响6,7,8,9,10,11.过渡金属催化中C-Si键活化的经典模式涉及产生离散的高价硅种12,13,14,15,16,17,18,19,从而促进了随后的跨金属化形成C-M中间体(图。1(A模式),硅部分最终作为副产物被去除。近年来,过渡金属氧化插入C-Si键的激活(图2)。1模式B)正引起越来越多的关注20.,21.在这种反应模式下,C-Si键断裂后形成新的Si-C键,这为C-Si键的功能化提供了机会,并创造了新的有机硅化合物,如各种硅环。然而,这样一个基本的步骤是非常具有挑战性的。目前C-Si键可以通过B模式激活的范围主要局限于环尺寸较小的硅氧烷22,23,24,25,26,27,28,29,30.,31,32,33,34,35,其中应变释放提供热力学驱动力。未应变Si−C(sp3.)由过渡金属插入形成的键仍然非常罕见。这些例子通常包括插入一个M−C(sp2)的物种一个变成Si−C(sp3.)结合,形成新的Si-C (sp2)以分子内方式结合,并专门生产硅土衍生物(图。1 b)36,37,38,39,40,41,42,43,从而限制了其合成的多功能性。我们想知道M−C(sp3.Si−C(sp3.)键,然后生成新的Si-C (sp3.)键,从而为不同的硅硅环提供了入口,而不是筒仓。

一个过渡金属催化中C-Si键裂解的两种一般活化模式。B文献及我们对无张力C(sp3.)-Si活化过渡金属插入。C我们的工作和可能的挑战在pd催化硅螺环化涉及C(sp3.-Si键通过插入Pd−C(sp3.)的物种。
受到C语言在应用方面的巨大进步的启发3.) -由烯烃分子内碳化产生的多米诺过程中的Pd种44,45,46,47,48,因此我们设计了烯系芳基碘化物1含三烷基硅基,验证了C(sp)的可能性3.) - Si键通过插入金属- C(sp3.)的物种。我们假设底物1要经过分子内5-外选择性碳钯化才能得到σ-烷基钯中间体吗Bβ-H缺乏,其反应活性可能与M - C相似2)的物种一个从而能够插入到C(sp3.) - Si键,然后还原消除生成新的C(sp3.−Si键(图;1 c).如果成功,该反应将成为构造结构独特的螺硅环的替代方法2.螺环支架在广泛的生物活性天然产物中被发现,并广泛应用于各种已批准的药物和催化剂中。尽管已有大量的快速获取螺环的方法,但催化制备螺硅环的方法仍然很少49由于C/Si开关在药物发现方面的广泛应用,受到了广泛的关注。然而,要实现我们提出的硅-螺旋体环化反应,可能需要解决以下挑战:(1)竞争性6-endo-selective Heck环化50;(2) σ-烷基钯中间体的or - c−H功能化B来传递螺旋熔合的苯并环丁烯4(无花果。1 c)51,52.
在本工作中,我们通过刻意调整反应条件,最大限度地减少副产物的生成,最终实现了pd催化的硅螺环化反应,包括连续的碳钯化,C(sp3.) -Si键解理/耦合。这项工作提供了一种方法,可以方便地以良好的产量获得不同的螺硅酸环。值得注意的是,如果(sp3.)通过插入M−C(sp3.)在我们的研究中观察到的物种可能为有机硅化合物的合成探索开辟机会。
结果与讨论
反应条件优化
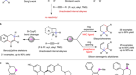
为了验证这个假设,我们决定采用1 aa39.该反应确实导致了所需要的螺旋体硅环的微量形成2 aa.为了显著提高收率,我们对一系列金属预催化剂、碱基、添加剂、配体和溶剂进行了详尽的筛选(见补充表)1).最后得到90%的分离收率2 aa是由(Pd)−1(5 mol%)作为(预)催化剂,LiOtBu (3.0 eq)作为碱,与AgOAc (2.0 eq)和铜(I)噻吩-2-羧酸(CuTc;0.2 eq)作为环己烷(0.4 M)的添加剂,在125°C下12小时(表1然后进行了一系列对照实验,以调查每个成分的影响。不出所料,在缺乏钯的情况下,没有理想的产物形成(条目2)tBu也是触发这种反应必不可少的(Entry 3)。我们推断I/Ot反应中间体中的Bu交换可能降低了反应活化能。CuTc的存在可能促进C(sp3.)-Si键裂解18,从而促进了5-外环化途径(Entry 4)。接下来,我们证明了醋酸离子对于提高催化剂的转化率至关重要,从而确保了高收率(Entries 5-8)。它会完全关闭反应,如果取代(Pd)−1由(Pd(烯丙基)Cl)2(条目9). Pd(Pt部3.)2能够以66%的产率引发反应(条目10)。这些结果表明,该反应可能由Pd(0)种引发。以甲苯为溶剂,反应收率达70%2 aa还有相当数量的6-内环化副产物3 aa(条目11)。最后,较低的催化剂负载量或较低的温度会阻碍其形成2 aa(第12及13项)。值得注意的是,理想产品的结构2 aa被单晶的x射线分析清楚地证实了。我们还成功地扩大了反应的规模,并在空气气氛下进行了反应1 aa作为底物。反应也顺利进行,没有显着的损失在收率(75%的收率在2.5 mmol;在空气气氛下获得80%的收率)。
底物范围
在优化条件下,我们接下来探索如图所示的衬底范围。2.取代基的范围R1在Ar的不同位置1首先对Ring进行评估。广泛的吸电子、电子中性和富电子取代基在c4位的耐受性良好,从而使所期望的螺硅酸环的产率适中或良好(2 ab−啊,收率48 ~ 95%)。c5取代底物在优化条件下反应良好,且产率无明显降低(2人工智能&2 aj,收率68-80%)。在c3位安装不同的取代基,会显著增加芳基碘化物的空间位阻,但不影响反应活性(2 ak党&2 al,收率分别为71%和73%)。在c6位置上承载- F或- Me基团的空间阻碍基板也顺利循环以产生所需的产物2点&2一个分别为69%和58%。密集取代的芳基碘化物也是有效底物,生成产物2 ak党−ao在58-73%的产量。的基于“增大化现实”技术1在螺旋硅环化反应中,环也可以是萘和异芳烃2美联社&2 aq.然后我们尝试改变R2-取代基在Ar上2环轴承的TMS基板组。我们首先证明,相对于TMS基团,在其元位或对位引入大量的吸电子、电子中性和富电子取代基并不会显著影响环化效率,从而提供了广泛的取代螺硅酸环(2 ba-bg),产量60 ~ 92%。与在Ar上有两个取代基的底物的反应2Ring也顺利地给出了相应的产品(2黑洞−bi).在TMS基团的相邻位置安装甲基并不妨碍所需产物的生成2 bj.在成功制备5,5-螺环之后,我们进一步证明了利用(2-碘苯基)甲醇衍生的底物,该螺硅环化方案也能产生5,6-螺环骨架。2 ca,收率45%)。值得一提的是,如果在Ar上的烯基上安装一个邻位取代基,就会完全阻碍螺旋环化反应,引发6-内环化2戒指。这可能是由于烯烃部分周围空间位阻增大所致。如果将底物中的氧系链原子改为氮系链原子,则不会对反应结果产生影响,并以中等产率得到相应的螺硅酸环。氮上的各种保护基团,如Ts、Ms和Ac,都与这种转化相容(2 da−直流).该过程成功地扩展到在Ar的4位和5位上含有卤素和其他电子中性、电子给电子和电子抽离取代基的基底上1环,提供了相应的螺硅酸环,产量良好(2弟弟−dh,收率40 ~ 70%)。

反应是用(Pd) 1(0.01 mmol), LiOtBu (0.6 mmol), AgOAc (0.4 mmol), CuTc (0.04 ~ 0.2 mmol)1(0.2 mmol)在环己烷(0.4 M)或甲苯(0.4 M)中浸泡4-12小时。孤立产量显示。
然后,在优化条件下,研究了硅中心不同取代基的影响(图。3.).在森那美2Bn和SiMe2等,空间受阻较小的Si - Me键优先裂解,得到相应的螺旋硅环2 ea(产量38%)和2 eb(68%收率),dr分别为1:1。产量低2 ea归因于SiMe ?位阻明显增大2Bn与经颅磁刺激组相比。的SiPhMe2取代基传递的混合物2 aa(产量23%)和2电子商务(产率22%),源于C(sp .2−Si键和C(sp3.)−Si键在反应下形成。此外,我们还证明了除了Si - Me键外,另一个C(sp3.)−Si键也能在该反应下裂解,SiEt反应证明了这一点3.提供2版60%的产量。收率的降低再次表明空间效应。

反应是用(Pd)−1(0.01 mmol), LiOtBu (0.6 mmol), AgOAc (0.4 mmol), CuTc (0.04 ~ 0.2 mmol)1(0.2 mmol)在环己烷(0.4 M)或甲苯(0.4 M)中浸泡4-12小时。孤立产量显示。产物的非对映比(dr)2由1H NMR。两种异构体均用柱状分离得到。
机械的研究
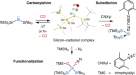
建立了结构多样化的螺旋体硅环文库2,我们首先证明了的保护基(Ts)2弟弟可以通过SmI治疗轻易去除2产生游离胺595%产率(图;4),从而进一步提高了该循环策略的综合效用。然后进行了几个对照实验,以了解反应机理。处理(Pd)−1在室温下,预催化剂在甲苯中加入AgOAc (2.0 eq)可生成新的Pd2复杂的(Pd) -OAc(无花果。4 b),已由单晶x射线晶体学证实。此外,就业率为5mol %(Pd) -OAc作为催化剂存在LiOt不,螺旋体硅环化反应1 aa高效地交付所需的产品2 aa80%产率(图;4 b).这些结果表明(Pd) -OAc在反应过程中可以形成络合物作为活性催化剂。此外,我们进行了反应1 aa在化学计量量的存在下(Pd) -OAc.大量的N,N-二甲基-1-苯基甲烷胺在反应过程中被GC-MS检测(见补充图)。1).另一方面,我们尝试在标准条件下通过气相色谱分析模型反应的气体组成来追踪裂解的Si-Me基团。我们发现该反应产生了相当数量的MeOtBu和MeI(图;4摄氏度).因此,我们相信我-/ tBuO-交换是在催化循环下发生的。

一个螺硅酸环中保护基团(-Ts)的去除2弟弟.B的合成、表征和应用(Pd) -OAc复杂。C气相色谱分析模型反应的气体组成。
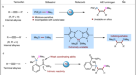
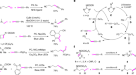
最后,进行了量子力学研究,对催化转化的基本步骤进行了进一步的机理研究,特别是对Pd催化剂的催化循环、C(sp)模式进行了研究3.-Si键的裂解和5-外环化高选择性形成所需C(sp3.) pd的物种。主要结果为反应的1 aa来2 aa见图。5.我们假设钯的催化量0在反应开始时原位形成,并与?的芳基- I部分氧化加成1 aa,产生Pd2-芳基中间体,与我们的实验结果一致(表1,分录10),XPS分析(见补充图)。2),并在文献中得到了充分的证明53,54.基于对可能的连接模式的评估,我们发现Pd2芳基物种IM12最稳定与的协调吗tBuO-(见补充图)3.而且4).的5-外环化IM12通过分子内迁移插入平稳发生(壹空间心肌梗死),提供C(sp3.) pd2物种IM22具有仅10.4千卡/摩尔的低屏障。相反,替代的6-内环化(壹空间的心肌梗死)由于桥接环结构的高应变,其势垒显著增大,为17.1 kcal/mol壹空间的心肌梗死,导致对所观察到的5-exo通路的强烈偏好。

自由能单位为千卡/摩尔,距离单位为Å。所有计算均在高斯16软件包中的SMD-PBE0-D3/def2-TZVP//M06-L/def2-SV(P)理论级别进行。
然后,我们将重点放在对Si-C (sp3.)键的裂解和随后的螺环化过程。尽管对各种初始几何进行了多次尝试,但我们无法找到一个假定的σ-methathesis过渡态。氧化加成过渡态TS2办公自动化而不是在所有收敛优化工作中得到。指出了三种Si-C (sp)中的一种的Pd(II)/Pd(IV)氧化加成过程3.)债券IM22通过过渡态TS2办公自动化实现Si-C (sp3.)裂解有利,生成Pd4-silametallacycle中间IM3455,56,57,58.然后,还原消去IM34随着新的Si-C (sp3.)键通过过渡态发生TS3再保险得到所需的螺硅酸环2 aa和Pd(II)中级IM42.迄今为止,这一途径的最大障碍是从IM22来TS2办公自动化该反应只需16.5千卡/摩尔,被认为是一种简单的反应。我们鉴定了原位生成的C(sp)的连续氧化加成/还原-消除过程3.) pd2物种是成功转化的关键机制基础。
形成的Pd(II)种IM42可以进一步还原消除以再生Pd(0)催化剂并完成催化循环或氧化加成芳基- I部分1 aa形成Pd(IV) -芳基中间体。进一步的DFT计算进行了比较这两种潜在的途径。从Pd(II)种还原消除IM42通过过渡态转变为Pd(0)种TS6再保险被计算出具有令人望而却步的39.6千卡/摩尔的高屏障。相反,氧化添加活性Pd(II)种IM42通过过渡态和芳基i反应TS4办公自动化形成Pd(IV)中间体IM54具有16.7千卡/摩尔的势垒和取代I的直接配体交换- - - - - -与tBuO- - - - - -在AgOAc和LiO在场的情况下t布鲁里溃疡。然后,进行随后的还原消去IM54通过过渡态发生TS5再保险生成MeO的能量势垒为24.8千卡/摩尔tBu和再生活跃的Pd(II)种IM12随着催化循环的完成。还原消去有利于Me-OtBu生成比Ar-Me键形成的动力学差1.2 kcal/mol,这解释了Ar-Me副产物的缺失(见补充图)。5).进一步分析还表明,I- - - - - -的结扎TS5”再保险而且TS5“RE都比tBuO- - - - - -协调TS5再保险,支持的稳定作用tBuO- - - - - -.总的来说,在Pd(II)/Pd(IV)流形上的催化系统的运行与我们的计算更符合,与实验证据吻合得很好。
综上所述,我们报道了pd催化的螺旋硅环化反应,该反应通过Heck反应/ Si−C(sp3.)解理/ Si−C(sp3.)键形成序列,允许构建不同的螺硅环。从机理的角度,我们的研究表明Si-C (sp3.)可以通过插入M−C(sp3.)的物种。这种反应模式可能为其他反应过程的发展提供机会。DFT计算表明:1)反应机制可能涉及Pd(II)/Pd(IV)催化循环;2) Si−C(sp3.σ-烷基钯(II)在Si - C(sp - C)上氧化加成生成了Pd(IV)3.)债券。
方法
一般程序
在一个充氮手套箱中,一个烤箱干燥的15ml螺旋盖密封管,用磁搅拌棒充电,1(0.20更易),(Pd) 1(5 mmol%), AgOAc (2 equiv), LiOtBu(3等),添加剂和环己烷(0.5 mL)或PhMe (0.5 mL)。将试管密封,然后从手套箱中取出,在125°C下,在N下搅拌形成的混合物212小时。冷却至室温后,饱和NH水溶液4加入Cl (5 mL),用EA (3 × 5 mL)提取。结合的有机相用水和盐水洗涤,干燥(MgSO4)然后蒸发了。采用反相柱(C18(ODS))制备反相高效液相色谱法纯化粗产物,洗脱液为CH3.CN)提供相应的产品。
数据可用性
作者声明,支持本研究结果的数据可在文章及其中获得补充信息文件,以及从相应的作者要求。计算结构的笛卡尔坐标可从补充数据中获得1.本研究中报道的结构的x射线晶体学坐标已在剑桥晶体学数据中心(CCDC)沉积,沉积编号为CCDC 2168893 (2 aa)及ccdc2168894 ((Pd) -OAc).这些数据可以通过剑桥晶体学数据中心免费获得www.ccdc.cam.ac.uk data_request / cif.
参考文献
王丽丽,段志忠。金属介导或催化Si-C键裂解形成硅氧烷。下巴。科学。公牛。58, 307-315(2013)。
Komiyama, T., Minami, Y. & Hiyama, T.过渡金属催化有机硅试剂合成转化的最新进展。ACS Catal。7, 631-651(2017)。
Bähr, S.薛,W. & Oestreich, M. C(sp3.)如果交叉耦合。ACS Catal。9, 16-24(2019)。
不对称催化中的硅立体硅烷。Synlett11, 1629-1643(2007)。
Fuchs, P. L.主编《有机合成试剂手册:硅介导的有机合成试剂》(Wiley: Chichester, UK, 2011)。
Bains, W. & Tacke, R.硅化学作为药物设计中化学多样性的新来源。咕咕叫。当今。药物。重击。6, 526-543(2003)。
肖维尔,G. A. &米尔斯,J. S.先导优化中的化学挑战:药物发现中的硅同位体。药物。今天8, 551-556(2003)。
Englebienne, P., Hoonacker, a.v. & Herst, c.v.生物等位硅取代在药物设计中的地位。毒品Des. Rev。2, 467-483(2005)。
弗朗茨,A. K. &威尔逊,S. O.有机硅分子与医学应用。医学。化学。56, 388-405(2013)。
Hirai, M., Tanaka, N., Sakai, M. & Yamaguchi, S.结构约束的硼-,氮-,硅-和磷中心多环π共轭体系。化学。牧师。119, 8291-8331(2019)。
克莱,H. F. T.等。水飞蓟离子:从难以捉摸的反应中间体到强有力的催化剂。化学。牧师。121, 5889-5985(2021)。
丹麦,S. E. & Regens, C. S.钯催化的有机硅烷醇及其盐的交叉偶联反应:硼和锡基方法的实用替代品。Acc。化学。Res。41, 1486-1499(2008)。
Nakao, Y. & Hiyama, T.硅基交叉偶联反应:一种环境友好的版本。化学。Soc。牧师。40, 4893-4901(2011)。
Foubelo, F, Nájera, C. & Yus, M. hiyama交叉偶联反应:新发现。化学。矩形。16, 2521-2533(2016)。
Minami, Y. & Hiyama, T.设计使用芳基(三烷基)硅烷的交叉偶联反应。化学。欧元。J。25, 391-399(2019)。
Rauf, W. & Brown, J. M.催化酰胺介导的甲基转移从硅烷到烯烃在藤原-森谷氧化偶联。Angew。化学。Int。艾德。47, 4228-4230(2008)。
Seiser, T. & Cramer, N.铑(I)催化的未活化硅烷从maryl到烷基的1,4-硅转移:吲哚醇衍生物的对映选择性合成。Angew。化学。Int。艾德。49, 10163-10167(2010)。
Nakao, Y., Takeda, M., Matsumoto, T. & Hiyama, T.通过烷基(三有机)硅烷分子内活化的交叉偶联反应。Angew。化学。Int。艾德。49, 4447-4450(2010)。
柯尔帕宁,S.,波利,G.,奥贝尔,J. &佩雷斯-卢娜,a.c (sp2)−醇氧化合物分子内活化的Si键功能化。欧元。j . Org。化学.2021, 1055-1071(2021)。
李玲,张勇,高林,宋哲。直接插入过渡金属活化C-Si键的研究进展。四面体。56, 1466-1473(2015)。
张庆林,安凯,何伟。Si-C (sp)催化合成π共轭硅硅烷3.)键激活。Synlett26, 1145-1152(2015)。
Franz, a.k. & Woerpel, k.a.硅环丙烷反应的发展,作为立体选择性有机合成的新方法。Acc。化学。Res。33, 813-820(2000)。
Hirano, K., Yorimitsu, H. & Oshima, K.镍催化与三烷基硼烷和硅环丁烷的反应。化学。Commun.3234 - 3241(2008)。
邮件用户代理,Q.-C。,Chen, J., Xia, C.-G. & Xu, L.-W. Synthesis of silacyclobutanes and their catalytic transformations enabled by transition-metal complexes.Coord化学。牧师。374, 93-113(2018)。
赵文涛,高峰,赵东。环丙烯与(苯并)硅环丁烷分子间σ-键交叉交换反应:硅(苯并)环庚酮的直接获得。Angew。化学。Int。艾德。57, 6329-6332(2018)。
陈,H.等。铑催化硅环丁烷与未活化的炔烃反应合成硅环己烯。Angew。化学。Int。艾德。58, 4695-4699(2019)。
冯,J.等。扭转应变促进rh催化芳基- narasaka酰化催化不对称C-Si键的活化。Commun Nat。11, 4449(2020)。
王,X.-B。et al。可控的Si-C键激活使钯催化的[4+2]环丙烯与苯并硅环丁烷环化成为可能。Angew。化学。Int。艾德。59, 790-797(2020)。
秦勇,韩建林,鞠春文,赵东。基于应变释放硅基交叉耦合的6-、7-和8元苯并硅环扩展。Angew。化学。Int。艾德。59, 8481-8485(2020)。
张,L.等。铑催化C-H硅化与硅环丁烷的结合计算和实验研究:导致更有效的催化剂体系的见解。j。化学。Soc。143, 3571-3582(2021)。
朱,M.-H。,Zhang, X.-W., Usman, M., Cong, H. & Liu, W.-B. PallAdium-catalyzed (4 + 4) Annulation Of Silacyclobutanes And 2-iodobiarenes To Eight-membered Silacycles via C–H and C–Si bond activation.ACS Catal。11, 5703-5708(2021)。
霍,等。钯催化的对映选择性碳宾插入硅环丁烷的碳硅键。j。化学。Soc。143, 12968-12973(2021)。
张娟,闫宁,琚长文,赵东。镍(0)催化不对称环膨胀生成富对映体硅立体生苯甲醚。Angew。化学。Int。艾德。60, 25723-25728(2021)。
Wang, W. et al. 3-硅氮杂环丁:一种尚未开发但用途广泛的有机硅烷物种,用于环向硅氮杂环的膨胀。j。化学。Soc。143, 11141-11151(2021)。
唐,等。硅环丁烷与烯酸酯的环膨胀,选择性地形成2-或3-(E)-烯酸取代的硅环己烯。ACS Catal。12, 5185-5196(2022)。
Ojima, I., Fracchiolla, D. A., Donovan, R. J. & Baneiji, P. 1,6-二炔的硅基碳自行车化:一种新的催化路线到双环[3.3.0]辛烯酮。j . Org。化学。59, 7594-7595(1994)。
tobiu, M., Onoe, M., Kita, Y. & Chatani, N.铑催化的2-硅基苯硼酸与炔的偶联导致苯并硅烷:三烷基硅基碳-硅键的催化裂解。j。化学。Soc。131, 7506-7507(2009)。
梁杨,张松,席哲。钯催化合成苯并西洛洛[2,3-b]吲哚的研究3.-Si键和相应的分子内C(sp2)如果耦合。j。化学。Soc。133, 9204-9207(2011)。
梁勇,耿伟,魏健,席哲。钯催化2-硅酰硅基溴与炔的分子间偶联:用C(sp)催化裂解合成苯并硅酮和异芳烃熔融硅酮3.如果债券。Angew。化学。Int。艾德。51, 1934-1937(2012)。
Onoe, M.等。铑催化碳硅键活化合成苯并硅衍生物。j。化学。Soc。134, 1947 - 19488(2012)。
RSC睡觉。3., 14273-14276(2013)。
张,Q.-W。,一个n,K. & He, W. Rhodium-catalyzed tandem cyclization/Si-C activation reaction forthe synthesis of siloles.Angew。化学。Int。艾德。53, 5667-5671(2014)。
杨,Q.等。铑催化炔烃分子内碳硅基化反应的研究3.) -Si键裂解。Org。化学。前面。5, 860-863(2018)。
弗拉尔,T.,鲁吉特,E. & Orru, R. V. A.钯催化级联环化的最新进展。放置合成器。Catal。353, 809-841(2011)。
3 - 7个C-C或c -杂原子键来自涉及赫克过程的多米诺反应。四面体69, 6735-6785(2013)。
KC, S. & Giri, R.结合碳金属化和交叉偶联对未活化烯烃的二碳功能化策略。j . Org。化学。83, 3013-3022(2018)。
杜嘉娜,李志强,李志强,王志强,王志强。过渡金属催化烯烃二碳功能化反应的研究。化学。矩形。18, 1314-1340(2018)。
李艳,朱建军,孔伟。钯催化碳钯化引发级联反应构建四元立体中心。Angew。化学。Int。艾德。58, 1562-1573(2019)。
徐毅,等。我3.SiSiMe2(OnBu):一种二硅烷试剂,用于通过Brook-和retrobrook -type重排合成各种硅氧烷。化学。科学。12, 11756-11761(2021)。
皮欧,李志强,朱晓明,朱晓明,等3.瞬态σ-烷基钯(II)配合物-H键:通过钯催化的多米诺碳钯化/C(sp3.) - c (sp3.)键形成过程。Angew。化学。Int。艾德。51, 11561-11565(2012)。
叶,J.等。由原位生成的钯环实现远端C - H烷基化和C - C键裂解。Nat,化学。9, 361-368(2017)。
叶,F.,葛,Y., Spannenberg, A., Neumann, H. & Beller, M.烯丙基铵盐在钯催化级联反应中合成螺熔杂环的作用。Commun Nat。11, 5383(2020)。
杨飞,张勇,郑荣,唐娟,何敏。叔芳胺环钯化配合物在Heck反应中的高效催化作用。j . Organomet。化学。651, 146-148(2002)。
Beletskaya, i.p.等人。nc -钯环是无磷化氢Heck反应的高效廉价前体。j . Organomet。化学。622, 89-96(2001)。
于志忠,兰勇。铑催化碳硅键裂解合成苯并硅衍生物的机理:计算研究。j . Org。化学。78, 11501-11507(2013)。
陈,W.-J。林卓。DFT对钯催化碳硅裂解合成苯并硅衍生物机理的研究。道尔顿反式。43, 11138-11144(2014)。
徐,L.-M。,Li, B.-J., Yang, Z. & Shi, Z.-J. Organopalladium(iv) chemistry.化学。Soc。牧师。39, 712-733(2010)。
Sehnal, P., Taylor, R. J. K. & Fairlamb, I. J. S.钯(IV)化学在合成和催化中的出现。化学。牧师。110, 824-889(2010)。
确认
感谢国家自然科学基金项目(22071114,22022103,21871146)、海河可持续化学转化实验室、国家重点发展计划项目(2019YFA0210500)、“新有机物质前沿科学中心”、南开大学(资助号63181206)、中央高校和南开大学基本科研业务费专项资金的支持。
作者信息
作者及隶属关系
贡献
y.s., x.s., j.z.和y.q进行了实验。B.L.进行DFT计算。D.Z.构想了这个概念,指导了这个项目,并撰写了论文。
相应的作者
道德声明
相互竞争的利益
作者声明没有利益竞争。
同行评审
同行评审信息
自然通讯感谢匿名审稿人对本工作的同行评审所作的贡献。
额外的信息
出版商的注意施普林格自然对出版的地图和机构从属关系中的管辖权主张保持中立。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议(Creative Commons Attribution 4.0 International License),允许以任何媒介或格式使用、分享、改编、分发和复制,只要您对原作者和来源给予适当的署名,提供知识共享许可协议的链接,并注明是否有更改。本文中的图像或其他第三方材料包含在文章的创作共用许可中,除非在材料的信用额度中另有说明。如果内容未包含在文章的创作共用许可协议中,并且您的预期使用不被法定法规所允许或超出了允许的使用范围,您将需要直接获得版权所有者的许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文

引用本文
施,杨,施,X,张,J。et al。硅螺环化涉及未张力C(sp3.−Si键断裂。Nat Commun13, 6697(2022)。https://doi.org/10.1038/s41467-022-34466-4
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/s41467-022-34466-4
