








摘要
我们首次提供了三个山兔亚种样本的全基因组序列。sylvaticus),以及两只山兔的样本(Lepus timidus timidus)和两只褐兔(天兔座europaeus)来自瑞典。与其他山兔(白色)和棕兔(大部分为棕色)相比,野野兔有独特的灰色冬季皮毛,并面临区域灭绝,可能是由于竞争排斥非本地棕兔。7只野兔标本的全基因组重测序被绘制到天兔座timidus假参考基因组,用于检测11,363,883个多态性核苷酸位置。这里提供的数据可能有助于解决瑞典山地兔和棕色兔的当地适应和保护状况,包括独特的亚种。
| 测量(s) | 全基因组测序 |
| 技术类型(年代) | Illumina HiSeq X |
| 因素类型(年代) | 物种 |
| 样本特征-有机体 | 小白兔•小白兔•小白兔 |
| 样本特征-位置 | 瑞典 |
背景与总结
在瑞典有两个形态上截然不同的山兔亚种,名义上的亚种(Lepus timidus timidus林奈,1758),广泛分布于北极/亚北极欧亚大陆,以及地方性的野野兔(sylvaticus尼尔森,1831)1这是一个更适合温带环境的亚种,分布在斯堪的纳维亚半岛南部和爱沙尼亚西部2.最明显的区别是它们冬季皮毛的颜色,名义上的山兔亚种是白色的,而野野兔是灰色的。
名义亚种的地理分布在瑞典北部已逐渐消退。其中一个可能的原因是来自引进的棕兔的竞争(天兔座europaeus帕拉斯,1778年)th世纪3.但它们的撤退也可能是由气候变化引起的伪装不匹配或其他栖息地的变化引起的4,5,6.虽然野野兔在无雪地区伪装得更好,但正如狩猎收获数据持续下降所表明的那样,由于栖息地改变和来自棕野兔的竞争,它也可能面临局部灭绝7.
早期的研究也表明了两个物种之间的种间基因流动(即渐渗),天兔座timidus而且天兔座europaeus8.渗透可以促进非本地棕色野兔适应北方的先决条件(例如季节性转移到白色冬季毛皮3.,7寒冷的气候和/或病原体抗性9).因此,这个两种物种的综合体为探索人类引起的生态变化如何快速影响物种,包括它们的行为、人口和进化相互作用,提供了一个极好的模型系统。除了上述的保护挑战之外,在瑞典,山兔和野野兔都是文化上重要的狩猎物种,数量的下降引起了人们的关注。
这里报告的基因组数据可用于解决复杂的适应模式,评估物种相互作用,并作为开发受威胁种群监测工具的资源。它们还为亚种差异和进化历史提供了有价值的见解。我们描述了全基因组重测序数据中的11,363,883个多态性,以检测差异标记,并提供了一个可能影响冬季毛皮颜色的色素沉着基因的示例分析筛选10.除了评估两种野兔的当地适应性、健康野兔的保护状况和开发管理工具外,所报告的基因组数据还可用于增加对整个分布的其他接触区山区野兔和棕兔之间相互作用性质的理解3.,7,11,12,13.结合收获数据、颗粒库存和相机捕获,这些具有潜在添加功能的基因数据可用于开发针对山地兔和褐兔的综合监测工具。
方法
样本采集和DNA提取
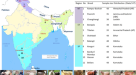
肌肉组织样本来自2005年至2017年瑞典常规狩猎季节拍摄的7只兔子。在瑞典南部,山地野兔的常规狩猎季节从9月1日持续到2月15日,在瑞典北部,狩猎季节一直持续到2月的最后一天。在瑞典,褐色野兔的常规狩猎季节是从9月1日到2月的最后一天。样本代表两种野兔和一种野兔亚种(表1)在瑞典(图。1):两个人天兔座europaeus,两个Lepus timidus timidus还有三个sylvaticus.所有三个sylvaticus(Le911, Le918和Le919)样本采集于1月和2月,此时蓝色/浅灰色冬季毛皮确认了亚种状态(表9)1;Angerbjörn & Flux 1995)。此外,样品从一个L. t.蒂米达斯(Le921)与l . europaeus在一月份采集,此时该亚种的白色冬季毛皮便于鉴定。最后三个样本的样本,一个L. t.蒂米达斯(Le922)和两个l . europaeus(LeA01和LeV01),分别采集于不同物种分布的地区(详情见表1).模板DNA按照制造商的说明使用QIAsymphony DNA Mini Kit提取。

(一个)褐兔样本分布图(l . europaeus;红)、石楠兔(L. timidus sylvaticus;青兔)及山兔(L. t.蒂米达斯;蓝色)。(B)变量的主成分分析。重叠点用黑色标出。(C)每个测序库的Phred质量评分。注意,Q30的phred分数相当于1000个基数中有1个不正确的基数,或者基数调用中99.9%的准确率。(D)按物种划分的共有和独特变异的维恩图。
文库制备和测序
这7个样品在Illumina HiSeqX的单个流池上的4个通道上测序。在瑞典斯德哥尔摩(Stockholm, Sweden)的sciilifelab (National Genomics Infrastructure Stockholm, Sweden)为每个兔样本准备了一个Illumina TruSeq PCR免费文库(Illumina, USA)。库的目标插入大小为350 bp。然后将样品以等摩尔比混合,并在Illumina Hiseq X平台上使用2 × 151 Hiseq X SBS化学试剂在4个通道上测序。使用Illumina软件包CASAVA的bcl2fastq_v2.19.1.403执行Bcl到FastQ的转换。
生物信息学
原始测序读数的质量14采用FastQC 0.11.915(无花果。1 c).然后使用cutadapt版本2.10从原始测序读取中删除适配器序列16接下来是进一步的阅读质量检查。然后将修剪后的读取映射到伪参考Lepus timidus基因组17(NCBI接入:GCA_009760805.1)与Burrows Wheeler Aligner, BWA版本0.7.1718的默认设置bwa mem算法。每个个体在不同的测序车道上进行测序,因此每个个体的所有文件都被用作Picard版本2.21.4 markduplduplicate的输入19步骤,每个个体得到一个单独的bam文件,去除PCR和光学重复。使用freebayes版本1.3.1在100,000 bp区域的基因组中调用变体20..VCFtools版本为0.1.1621用于对原始变量文件进行四轮变量过滤。首先,我们允许最大缺失基因型率为50%,最小等位基因数为3,最小变异phred质量评分为30。其次,我们应用了一个较小的等位基因频率截断值0.05和最小平均深度20。第三,我们筛选等位基因平衡(AB)的变体:AB > 0.25 & AB < 0.75 | AB < 0.01。最后,应用了一个允许最大平均深度为30的硬过滤器。这种过滤产生了具有高质量调用的变体文件。命令和工具的详细列表可以在表中找到2.最终VCF文件中的基因型分期是使用beagle 4.1版本实现的22.
最终VCF文件的详细信息23根据样本种或亚种对VCF文件进行划分,计算共享和唯一snp的数量。然后使用vcffixup 1.0.0版本重新计算变量统计数据,并过滤以只保留变量位点(AC > 2和AF < 1)。23.最后,将私有VCF文件转换为床文件,并利用干涉算法计算床文件的交点24.R版本4.0.4估计种群分化(2021-02-15)25使用glpca函数包含在包adegenet 2.1.5版中26,27.
为了生成与其他野兔物种的线粒体系统发育树,我们组装了每个个体的线粒体基因组。使用NovoPlasty 4.3版本组装线粒体基因组28.NovoPlasty需要一个种子序列来构建组装,因此我们使用了整个线粒体基因组天兔座timidus(Genbank登录NC024040)作为种子序列。然后将组装好的线粒体基因组与小鼠的基因组进行比对Oryctolagus cuniculus(注册编号AJ001588.1),l . capensis(注册编号GU937113.1),l .也(注册编号NC_024043.1和KJ397613.1),l . townsendii(注册编号NC_024041.1和KJ397609.1),l . coreanus(注册编号KF040450.1),l . granatensis(注册编号NC_024042.1, KJ397610.1),l . tolai(注册编号KM609214.1),l . yarkandensis(注册编号MN450151.1和MG279351.1),l . sinensis(注册号KM362831.1),l . hainanus(注册编号JQ219662.1),l . capensis(注册编号GU937113.1),l分析(注册编号LC073697.1),l . europaeus(登录号AJ421471.1和KY211025.1)l . timidus(登录号NC_024040.1, KJ397605.1, KR030072.1, KR030070.1, KR030069.1, KR013248.1和KR019013.1)使用MAFFT v 7.49029.然后使用智商树web服务构建系统发育树30.,由ModelFinder确定最佳模型31并使用2000次超快速自举迭代。第二种系统发育树是利用已确定的全基因组变异生成的。系统发育树估计为RaXML32版本8.2.12,带有GTR + Γ + ASC模型和200个快速自举复制。然后在Ape 5.4版本中使用chronos功能对整个基因组ML树进行超测量33的估计散度时间天兔座europaeus而且天兔座timidus.
验证
使用FastQC对测序质量进行技术验证,然后使用MultiQC编译数据34.为了验证数据集的来源,我们使用了两种方法,首先确认冬季表型,其次确认系统发育关系及其祖先系数。系统发育关系是根据上述包括其他野兔物种在内的线粒体基因组进行评估的,然后使用来自封闭分析的全基因组变异。使用Poppr版本2.9.3确定祖先系数35基于DAPC方法。
使用的例子
为了进一步说明数据集的潜在用途,我们对59个已知色素沉着基因中的snp进行了示例分析10,这可以用来了解皮毛颜色的遗传学(图。3.).首先,下载表中每个基因的兔或小鼠参考序列2Hoekstra,(2006)从NCBI数据库。然后这些基因被映射到伪参考Lepus timidus基因组使用minimap236拼接感知转录算法。然后,我们计算了基于BEDtools映射所识别的皮毛颜色基因区域重叠的snp的数量37并给出该数据(图;3.).
数据记录
测序数据
测序是成功的,结果总共有32.4亿次读取。测序阅读质量较高,平均phred值大于Q30(图3)。1 c).对象的平均映射速率假参考Lepus timidus93.59%,平均79.9%的reads映射为合适的配对(表2)3.).原始测序数据14可以从NCBI短读档案下的项目登录和个人登录号码可以在表1.
变量调用和过滤
变体调用是在CSC Puhti服务器上使用freebayes软件的并行版本执行的2(CSC -科技中心,芬兰)。Freebayes是一个单倍型敏感的变体调用者,在整个基因组中一共识别了78,968,182个变体。应用各种过滤器后(表2),我们回收了11,363,883个高质量变异(表4)分布在整个假参考基因组中。
线粒体基因组组装
我们为每个个体组装了接近完整的线粒体基因组序列。线粒体基因组组装长度从16,648 bp到17,612 bps,有4个个体产生环状contigs (LeA01, LE911, LE919和LE921)。所有线粒体基因都存在于每个个体中。在本研究中产生的线粒体基因组的对齐包括作为phylip格式的Dryad数据集的一部分23.
技术验证
主成分分析(图;1 b)和complot(图;2摄氏度),进一步证实了不同的物种分组。类似的分组模式已在天兔座在芬兰各地取样的物种38的遗传多样性l . europaeus较小,可能是由于方正效应导致这些样本的紧密聚类,而更广泛的聚类l . timidus样品(图。1 b).种间渗透也可能影响观察到的模式38.在11,363,883个高质量的变种中,1,23,035个是独一无二的l . europaeus596,291到l . timidus(无花果。1 c).

(一个)基于样本全基因组单核苷酸变异的超精细系统发育树。报告节点支持<100。系统发育被校准到间隔大约300万年l . europaeus而且l . timidus41.(B)几种线粒体全基因组的极大似然树天兔座物种。这棵树已长了根Oryctolagus cuniculus.小费标签的颜色根据样品收集的国家而定。报告节点支持<100。(C)野兔种群的复图分析,显示每个野兔样本的后验隶属度概率。注意两个山兔亚种之间的区别。
为了确认野兔样本之间的关系,我们基于全基因组snp生成了最大似然(ML)系统进化树(图2)。2).这些样本的系统发育聚类证实了样本的起源,因为第一个分支分离了天兔座europaeus而且天兔座timidus,其次是分离的l . timidus亚种L. timidus sylvaticus.该亚种在来自大陆的样本和瑞典西南部岛屿上收集的样本之间产生了进一步的分裂。同样,基于线粒体基因组的系统发育(图。2 b),首先显示l . europaeus而且l . timidus样本。然后L. timidus而且L. timidus sylvaticus样品被进一步分割。有趣的是,还有其他的l . timidus来自远东的基因组L. timidus sylvaticus与这些样本聚在一起。观察到的全基因组和线粒体基因组数据之间的差异可能受到渐渗模式以及不同的遗传方式的影响。
使用笔记
所描述的基因组数据可用于评估野兔在局部、区域或大范围分布中的适应性,例如色素沉着、耐寒性和病原体抗性。一个特别的价值是开发一个综合监测工具,以评估瑞典南部和中部的野野兔的保护状况。这可以通过整合基因数据、收获数据、颗粒库存和摄像机捕获来实现。除了一般的保护之外,从资源的角度来看,长期保护野野兔,以及理解山兔和棕兔之间相互作用的本质是很重要的,因为野兔在瑞典和其他地方是一种狩猎物种,因此是肉类和娱乐的来源。
尽管样本集很小,数据表明在两个山兔亚种中有显著数量的私有等位基因(图2)。1 d).由此导致的两个演化支之间的分歧(图。2 a, B)在系统发育和祖先合集分析中清晰可见(图。2摄氏度),支持亚种代表生物实体,而不仅仅是单一基因的冬季毛皮颜色差异39.此外,这些私有等位基因可用于通过SNP面板检测野兔及其亚种的多样化。亚种在无花果中分裂。2可能从最后一个冰期末期开始就一直保持着同域关系40尽管时间还需要进一步验证40.维持遗传分裂的机制值得进一步研究,并提出了一个关于遗传分裂的有趣问题善意的野野兔的亚种状况。
为了说明如何将这些基因组数据与未来的分析相结合,我们计算了在已知色素沉着基因中发现的变异的数量。虽然,这项分析的样本量本身太小,无法提供任何结论,但我们能够识别出基因内部和周围的一些变异(图2)。3.),对色素沉着有已知影响10,39.如果与更大的数据集集成,我们相信这里提供的基因组数据将为种群遗传学、保护生物学和进化研究提供有价值的资源。

单核苷酸变异的数量及其周围的已知基因负责的皮毛颜色在小鼠。
代码的可用性
所有用于显示这些数据的程序和命令都包含在Table中2.
参考文献
Angerbjörn, A. & Flux, J. E. C. Lepus timidus。Mamm。物种1 - 11,https://doi.org/10.2307/3504302(1995)。
关于北极野兔的一个独特种群——健康野兔的遗传学、进化和分布历史,天兔座timidus林。Swed。Wildl。(Viltrevy)6, 381-460(1969)。
Thulin C.-G。山兔的分布天兔座timidus在欧洲:来自褐兔L. europaeus的挑战?Mamm。牧师。33, 29-42(2003)。
米尔斯,L. S。等.由于雪持续时间减少,迷彩在季节外套颜色上不匹配。Proc, Nat.Acad。科学。1107360-7365(2013)。
Zimova, M。等.物候变化的缺乏导致山地兔伪装不匹配的增加。Proc.Royal Soc。B:生物。科学。287, 20201786(2020)。
Levänen, R., Kunnasranta, M. & Pohjoismäki, J.线粒体DNA在褐兔北部边缘的渐渗(天兔座europaeus)范围内。Ann Zool Fennici55, 15-24(2018)。
Thulin C.-G。,Winiger, A., Tallian, A. G. & Kindberg, J. Hunting harvest data in Sweden indicate precipitous decline in the native mountain hare subspeciessylvaticus(健康兔)。纳特。64, 126069(2021)。
Thulin C.-G。,Jaarola, M. & Tegelström, H. The occurrence of mountain hare mitochondrial DNA in wild brown hares.摩尔。生态。6, 463-467(1997)。
Pohjoismäki, J. L. O.,米歇尔,C., Levänen, R. &史密斯,S.与山兔杂交增加褐兔的功能性等位基因库。科学。代表。11, 15771(2021)。
遗传,发展和进化的适应性色素沉着在脊椎动物。遗传(Edinb)97, 222-234(2006)。
哈米尔,R. M. Doyle, D. & Duke, E. J.欧洲山兔亚种遗传多样性的空间格局,天兔座timidusl遗传(Edinb)97, 355-365(2006)。
Leach, K, Montgomery, W. I. & Reid, N.生物地理学、宏观生态学和物种特征在Lagomorpha目中介导竞争相互作用。Mamm。牧师。45, 88-102(2015)。
马奎斯,j.p.。等.数据描述:山兔转录组和诊断标记作为监测与欧洲兔杂交的资源。科学。数据4, 1-11 (2017)
序列读存档https://identifiers.org/insdc.sra:SRP358660(2022)。
FastQC:高通量序列数据的质量控制工具。Babraham生物信息学.预印在http://www.bioinformatics.babraham.ac.uk/projects/fastqc(2010)。
Martin, M. Cutadapt从高通量测序读取中移除适配器序列。EMBnet.journal17, 10-12(2011)。
马奎斯,j.p.。等.山兔基因组注释草图(天兔座timidus).基因组医学杂志。另一个星球。12, 3656-3662(2020)。
李,H. & Durbin R.快速和准确的短读对齐与Burrows-Wheeler变换。生物信息学25, 1754-1760(2009)。
广泛的研究所。皮卡德工具包。Broad研究所,GitHub仓库.预印在https://broadinstitute.github.io/picard/(2019)。
Garrison, E. & Marth, G.基于单倍型的短读测序变异检测。arXiv 1207.3907(2012)。
Danecek, P。等.变量调用格式和VCFtools。生物信息学27, 2156-2158(2011)。
Browning, S. R. & Browning, B. L.利用局部单倍型聚类进行全基因组关联研究的快速和准确的单倍型分期和缺失数据推断。点。j .的嗡嗡声。麝猫。81, 1084-1097(2007)。
米歇尔,C. T., Pohjoismäki, J. L. O.,斯庞,G. & Thulin, C.-G.;利用山兔和褐兔的遗传多态性来调查野野兔的本地适应和保护状况(sylvaticus(Nilsson 1831)。德律阿得斯,https://doi.org/10.5061/dryad.3bk3j9kmp(2022)。
Khan, a . & Mathelier, a . Intervene:用于多个基因或基因组区域集合的交叉和可视化的工具。BMC生物信息学18, 287(2017)。
R核心团队。R:用于统计计算的语言和环境。R统计计算基金会,维也纳,奥地利.http://www.R-project.org/.
Jombart, T. & Ahmed, I. adegenet 1.3-1:全基因组SNP数据分析的新工具。生物信息学27, 3070-3071(2011)。
Jombart, T. adegenet:用于遗传标记的多元分析的R包。生物信息学24, 1403-1405(2008)。
Dierckxsens, N., Mardulyn, P. & Smits, G. novo成形术:从全基因组数据重新组装细胞器基因组。核酸测定.45(2017)。
Katoh, K. & Standley, D. M. MAFFT多序列比对软件版本7:性能和可用性的改进。摩尔。杂志。另一个星球.30.(2013)。
Trifinopoulos, J., Nguyen, L. T., von Haeseler, a . & Minh, B. Q. W-IQ-TREE:用于最大似然分析的快速在线系统发育工具。核酸测定.44(2016)。
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder:用于精确系统发育估计的快速模型选择。Nat方法。14, 587-589(2017)。
Stamatakis, a . RaxML版本8:用于系统发育分析和大系统发育后分析的工具。生物信息学30., 1312-1313(2014)。
帕拉迪斯,E. & Schliep, K.猿5.0:现代系统发育和进化分析的环境R。生物信息学35, 526-528(2019)。
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC:在一份报告中总结多种工具和样品的分析结果。生物信息学32, 3047-3048(2016)。
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr:克隆、部分克隆和/或有性生殖群体遗传分析的R包。PeerJ2, e281(2014)。
李,H. Minimap2:核苷酸序列的成对比对。生物信息学34, 3094-3100(2018)。
Quinlan, a.r. & Hall, i.m. BEDTools:一套用于比较基因组特征的灵活工具。生物信息学26, 841-842(2010)。
Levänen, R.,苏林,C.-G.;,Spong, G. & Pohjoismäki, J. L. O. Widespread introgression of mountain hare genes into Fennoscandian brown hare populations.《公共科学图书馆•综合》13, e0191790(2018)。
Giska,我。等.山地兔局部适应的进化途径。摩尔。生态。31, 1487-1503(2022)。
Thulin C.-G。,Isaksson, M. & Tegelström, H. The origin of Scandinavian mountain hares (天兔座timidus).Gibier Faune Savage/游戏与野生动物14, 463-475(1997)。
费雷拉,m.s。等.兔子在辐射过程中反复渗入的后遗症。系统。医学杂志。70, 593-607(2021)。
确认
c.g.t.感谢瑞典环境保护署(项目编号802-0216-15和NV-05813-21)的财政支持,并协助从Bo Gunnarsson、Anders Friberg、Martin Hahne和Jonas Malmsten的野兔中采集样本。
作者信息
作者及隶属关系
贡献
Craig T. michelle:生物信息学方法论、调查、形式化分析、可视化和写作(初稿、评审和编辑)。Jaakko L. O. Pohjoismäki:生物信息学管道设计、调查、监督和写作(审查和编辑)。Göran Spong:概念化、方法论、调查和写作(回顾和编辑)。Carl-Gustaf Thulin:概念化,实验设计和写作(初稿,评审和编辑)。
相应的作者
道德声明
相互竞争的利益
作者声明没有利益竞争。
额外的信息
出版商的注意施普林格自然对出版的地图和机构从属关系中的管辖权主张保持中立。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议(Creative Commons Attribution 4.0 International License),允许以任何媒介或格式使用、分享、改编、分发和复制,只要您对原作者和来源给予适当的署名,提供知识共享许可协议的链接,并注明是否有更改。本文中的图像或其他第三方材料包含在文章的创作共用许可中,除非在材料的信用额度中另有说明。如果内容未包含在文章的创作共用许可协议中,并且您的预期使用不被法定法规所允许或超出了允许的使用范围,您将需要直接获得版权所有者的许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文

引用本文
米歇尔,C.T, Pohjoismäki, J.L.O, Spong, G。et al。利用山兔和褐兔的遗传多态性来调查野野兔的本地适应和保护状况(sylvaticus(Nilsson 1831)。科学数据9, 667(2022)。https://doi.org/10.1038/s41597-022-01794-5
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/s41597-022-01794-5
