





摘要
Nibea coibor海蜇属海蜇科,分布于南海、东海、印度、菲律宾等海域。在这项研究中,我们对一个男性的DNA进行了测序Nibea coibor使用PacBio长读测序并生成染色质相互作用数据。基因组的大小Nibea coibor根据海蜇生成的k-mer计数估计为611.85~633.88 Mb。PacBio测序产生了29.26 Gb的HiFi reads, Hifiasm用于组装627.60 Mb的基因组,contig N50为10.66 Mb。我们进一步发现,24条染色体的端粒均存在典型端粒重复“TTAGGG”。使用BUSCO和merquury,组装的完整性分别估计为98.9%和97.8%。使用的组合从头开始通过预测、蛋白同源性和RNAseq注释,共鉴定出21433个蛋白编码基因。系统发育分析表明Nibea coibor而且Nibea albiflora密切相关。研究结果为植物遗传育种和基因组进化研究提供了重要依据Nibea coibor.
| 测量(s) | 全基因组测序•转录谱分析 |
| 技术类型(年代) | 单分子实时测序•RNA测序 |
| 样本特征-有机体 | Nibea coibor |
| 样本特征-位置 | 中国 |
背景与总结
Nibea coibor海蜇属海蜇科,主要分布在南海、东海、印度、菲律宾等海域1(无花果。1).它是一种快速生长的鱼类,在中国沿海广泛养殖,具有很高的营养价值和经济价值。早期对这种鱼的研究主要集中在养殖方法和生物学特性方面。近年来,研究主要集中在饲料营养方面2,3.,4,5,6,经济增长7,8,9和发展10,11,12.有关于线粒体基因组的报道Nibea coibor1,13;然而,缺乏基因组组装阻碍了对该物种的遗传和进化研究。

的照片Nibea coibor.
最近,单分子测序14由于其读取长度长、速度快、准确度高的优点发展迅速,已成为基因组组装的主流测序方法。该技术已成功应用于鱼类基因组的组装,如Oreochromis mossambicus15,Acanthopagrus边16,Scatophagus argus17而且Hypophthalmichthys molitrix18.PacBio最新更新的在循环一致测序(CCS)模式下产生的高保真(HiFi)序列读取实现了读取长度和碱基质量之间的平衡19.一些用于处理HiFi读取的汇编软件,包括HiCanu20.,猎鹰21和Hifiasm22,可用。其中,Hifiasm22是用于长HiFi读取的最新单倍型解析基因组组装算法。Hifiasm首先执行全对全读重叠比对,然后在默认情况下对排序错误执行三轮错误纠正。然后使用修正后的读数再次生成重叠对齐并构建字符串图。如果存在杂合等位基因,Hifiasm任意选择一个单倍体,并输出一个主装配和一个备用装配。它可以解析重复的序列信息,如着丝粒和端粒信息。与现有的其他算法相比,Hifiasm22具有装配速度快、精度高、连续性好等优点。长长的高保真序列读出了Hifiasm22装配算法,结合Hi-C23技术,实现高质量的染色体级基因组组装。然而,Hifiasm不能正确地解析高度重复的区域24.
在这项研究中,我们从一名男性身上提取了DNANibea coibor并使用PacBio平台生成HiFi读取。使用Hifiasm生产了高质量的contig组件。与Hi-C数据一起,Juicer和3D-DNA被用于组装和生成染色体水平的基因组。然后使用三种策略来注释基因组。此外,还进行了基于单拷贝基因的系统发育分析,以了解它们之间的关系Nibea coibor还有其他物种。这是第一个基因组组装Nibea coibor这将有助于了解该物种的基因结构、功能和排列,为后续的遗传育种、进化分析和种质资源保护研究提供依据。
方法
图书馆建设与排序
基因组DNA是从一个男性的肝脏和鳍中分离出来的Nibea coibor分别采用苯酚/氯仿法进行长读测序和短读测序。HiFi SMRTbell库使用SMRTbell Express Template Prep Kit 2.0 (PacBio, CA, USA)制备。用g-TUBE (Covaris, MA, USA)将gDNA剪切至15~18 kb,使用Template Prep Kit中的试剂修复DNA损伤和片段末端。将SMRTbell发夹接头连接到修复的末端,然后使用AMPure PB珠(PacBio, CA, USA)进行文库浓缩和纯化。为了获得用于测序的大插入SMRTbell库,使用BluePippin系统选择大于15 kb的SMRTbell模板(SageScience, MA, USA)。测序由Novogene(北京,中国)公司使用PacBio Sequel II平台进行。随后,CCS软件(https://github.com/PacificBiosciences/ccs)用于生产质量在Q20以上的高精度HiFi读数,标准设置为Min passes = 3, Min RQ = 0.99(表1).使用cutadapt (v2.10)检查了HiFi读取中的SMRTbell适配器污染25,要求与适配器序列至少有15 bp的重叠(错误率= 0.1)。我们发现1919,461个读取中只有284个包含适配器,并且被适配器污染的读取被过滤掉了。最后,我们保留了29.26 Gb的HiFi数据,读取长度的最长长度、平均长度和N50分别为39.74 kb、15.24 kb和15.34 kb2),分别。Novogene (Beijing, China)使用Illumina NovaSeq 6000平台对从鱼鳍中提取的DNA进行测序,生成19.79 Gb原始配对端reads,读取长度为150 bp。
根据制造商的说明,使用TRIzol试剂(Invitrogen, MA, USA)从一名男性和一名女性的肝脏、肌肉、睾丸和卵巢组织中提取总RNA,然后以等摩尔浓度聚集用于RNA测序。用寡聚(dT)珠筛选总RNA,加入裂解缓冲液将总RNA裂解成短片段。这些短片段用随机六聚体引物合成第一链cDNA,然后合成第二链cDNA。使用AMPure XP珠纯化双链cDNA,使用EB缓冲液进行末端修复和a尾。对构建的RNA文库进行定量和稀释,并使用Agilent 2100生物分析仪系统(Agilent Technologies, CA, USA)评估插入物的大小。qPCR法准确定量文库有效浓度。使用Illumina NovaSeq 6000平台(Novogene, Beijing, China)对RNA文库进行测序,共获得17.04 Gb的配对端原始reads, Q30为93.67%(表3)1).
Hi-C数据来自一名男性的肝组织样本Nibea coibor.Hi-C库是按照Belton所描述的方案使用肝组织构建的等.26,做了一些修改。简而言之,将组织研磨,然后与4%的甲醛溶液交联。将核重悬于NEB缓冲液中,用稀SDS增溶,用4切酶MboI(400单位)进行酶切。用苯酚-氯仿萃取纯化DNA。利用Illumina NovaSeq 6000平台对构建的文库进行配对端测序。对排序后的原始数据进行过滤,得到总共88.96 Gb的干净数据(表1), Q20 = 96.74%, Q30 = 91.82%,用于辅助染色体组装。
组装和基因组质量评估
使用Hifiasm (v0.13.0-R307)的默认参数组装基因组22.我们使用没有附加数据(如父代数据)的HiFi读取来生成主组装图。我们预先计算了重叠,并从修正后的读取中重新执行重叠,用Hifiasm清除了haplotig重复,并进行了三轮错误修正。生成314个contigs,大小为627.60 Mb。最大contig大小为23.26 Mb,最大N50为10.66 Mb2),分别。
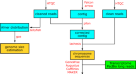
榨汁机27(v1.6)结合3D-DNA28(v180419)用于脚手架。首先,HiCUP29(v0.8.1)对Hi-C数据进行处理。BWA30.(v0.7.17-r1188)用于contig水平基因组的索引,然后使用Juicer创建限制性内切酶切割位点。利用Juicer (v1.6)对处理后的Hi-C数据进行进一步分析和处理。简而言之,我们设置了限制性内切类型(S)、参考基因组文件(Z)、限制性内切酶切割位点文件(Y)和染色体大小文件(P)。利用3D-DNA的run- asm -流水线.sh脚本构建参考基因组草图,并使用3D-DNA生成组装热图(图2)。2).Juicerbox31(v1.11.08)用于手动纠正装配错误(主要是易位错误),最终我们解决了24条染色体(图。3.).3D-DNA的run-ASM-pipeline-post-review.sh脚本28再次使用Juicerbox对修改后的文件输出结果进行修正,得到“FINAL”装配,共230个支架。支架最大尺寸为31.60 Mb, N50最大尺寸为26.22 Mb2),分别。

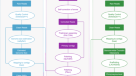
基因组组装、注释和系统发育的工作流程。

的全基因组Hi-C热图Nibea coibor.蓝色方块代表染色体蓝色方块内的绿色小方块代表染色体的contigs。灰色区域所包含的蓝色方块是弹片。
此外,基于脊椎动物端粒序列信息检测组装基因组中端粒重复序列的分布32由端粒酶资料库提供(http://telomerase.asu.edu/sequences_telomere.html).结果显示,24条染色体均含有端粒重复序列,即重复序列' TTAGGG '及其反向补体' CCCTAA ',其中14条染色体含有大量的重复序列,从14到1365不等(补充图)。1).
基因组大小和完整性估计
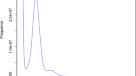
水母33(v2.3.0)通过设置k-mer参数为19、23、27和31来计数k-mer(表2.3.0)3.和补充图。2),并利用高覆盖短读得到相应的频率分布。的估计基因组大小Nibea coibor从611.85 Mb(19个月)到633.88 Mb(23个月)3.,补充图。2).
基准通用单副本正交镜(BUSCO)34(v5.1.2)还用于评估actinopteryGIi_ODb10数据库的基因组完整性(https://busco-data.ezlab.org).共鉴定出3,640个BUSCO基因,其中完整基因3,600个,单拷贝基因3,552个,多拷贝基因48个,缺失基因29个,分别占全基因组的98.9%、97.6%、1.3%和0.3%(表4).此外,墨丘利35使用HiFi和Illumina reads来评估基因组的QV值和完整性。结果,使用HiFi和Illumina短读,基因组的完整性达到97.8%。用HiFi和Illumina k-mers估计qv分别为61.9和46.6。用merquery生成的k-mer谱图显示,在我们的基因组组装中没有异常的虚假重复,并且k-mer只出现在组装中,而不出现在测序reads中(意味着组装中的碱基错误),是微不足道的(补充图。3.).
重复‐内容识别和注释
RepbaseTE文库使用RepeatMasker程序检测染色体尺度基因组组装中的重复序列36(v4.0.6)和RepeatModeler37(v1.0.9)用于构造从头重复库。结果显示,重复序列为11.49 Mb,占组装基因组的18.31%。在重复序列元件中,短点缀核元件(sin)占基因组大小的0.58%,长点缀核元件(LINEs)占基因组大小的1.79%。长端重复序列(lts)和DNA元件分别占1.37%和3.11%。小RNA含量为0.46%,卫星和简单重复序列分别占0.15%和2.72%。
综合策略:从头开始采用转录证据和基于蛋白质同源性的基因预测方法进行基因注释。汇集的RNAseq清洁数据通过两种方式进行组装,即依赖于参考基因组的转录本组装和使用Trinity软件进行de novo组装38(v2.4.0)和开放阅读帧(orf)使用PASA进行识别39(v2.1.0)。奥古斯都40(v3.2.3)执行从头开始利用已知的斑马鱼基因和RNAseq转录本进行基因预测。经过两轮模型训练,得到最优参数。Tblastn41被用来排列蛋白质序列Nibea coibor还有其他9个物种,包括Cynoglossus semilaevis, Danio rerio(斑马鱼),Takifugu rubripe(河豚),Dicentrarchus labrax(欧洲鲈鱼),Gasterosteus aculeatus(三个量有脊柱的棘鱼), Larimichthys鳄鱼(大黄鱼),晚期钙化动物,尼罗岩石层而且Oryzias latipes(medaka),用于基于同源的基因预测。天灾42(v2.2.0)用于精确定位对齐序列的剪接位点和外显子。剔除编码区域小于150bp的基因,利用证据建模器(Evidence modeler, EVM)对三种基因预测模型的结果进行加权和评价。43(v1.1.1)以产生包含编码区域和可选剪接位点的全面可靠的基因结构。所有预测基因均与NCBI非冗余蛋白(nr)数据库进行比对,并使用blastp进行功能注释44.最终,预测了21433个基因,包括14633个非选择性剪接基因和6800个选择性剪接基因。在这些基因中,有19859个基因在NCBI nr数据库中进行了注释。
系统发育分析
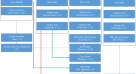
13个物种的编码序列(CDSs),包括智人,波达西muralis, Gallus,眼细尾鱼,大鲵,鳄尾鱼,斑纹剑鱼,黑绿四齿兽,尼罗裂唇鱼,墨西哥稻谷,针叶棘齿鱼,白花尼贝鱼45而且Collichthys光泽的46,是从Ensmbl或NCBI数据库中检索的。提取各种各基因的最长CDS,利用OrthoFinder进行同源性分析47(v2.5.4)使用默认设置。14个物种共鉴定出333,401个基因,其中同源单拷贝基因1,876个。使用Muscle的-align参数对这些同源单拷贝基因进行比较48(v5.1)。Gblock49,50(v0.19b)使用参数“-b4 = 5 -b5 = h -t = d -e = 0.2”提取比较结果中的保守序列,Seqkit51(v2.2.0)用于合并结果。通过MEGA11构建系统发育树52,与智人有关作为外群,和时间树53以鸡和蜥蜴的分化时间(280 MYA)为基础,估计其他脊椎动物的分化时间。利用iTOL对进化树进行可视化54(https://itol.embl.de/).根据我们的系统发育树(图。4),我们观察到Nibea coibor进化上更接近吗Nibea albiflora,也属于Nibea,散度时间为16.9 MYA。此外,这两个物种有一个共同的祖先Larimichthys稚鱼而且Collichthys光泽的,属于同一科Sciaenidae,两个分支的分化时间为26.4 MYA。

的系统发育分析Nibea coibor和其他13个物种。分支上的绿色数字表示每个位点核苷酸取代的平均数量(分支的长度不能准确代表取代率),节点附近的黑色数字表示分化时间(百万年前,MYA),括号内的蓝色数字是bootstrap值。
线粒体的完整序列(GenBank ID: CM041792.1)Nibea coibor包含在我们的程序集中。线粒体包含13个蛋白质编码基因,22个tRNA和2个rRNA基因,用MITOS Web Server注释55(http://mitos.bioinf.uni-leipzig.de/index.py).在上述13个物种中,线粒体CDSs最长Nibea coibor使用Clustal Omega (v1.2.4)进行比较56.利用IQ-TREE (v1.6.12)构建基于线粒体序列的系统发育树57,58他认为Nibea coibor更接近于Nibea albiflora,Larimichthys稚鱼而且Collichthys光泽的(补充图。4).
技术验证
使用琼脂糖凝胶电泳检查提取的DNA对端测序,并使用量子位荧光仪(Thermo Fisher Scientific, USA)测定DNA浓度。
用琼脂糖凝胶电泳检测PacBio测序提取的DNA,主条带大于30 kb。DNA浓度使用量子位荧光仪(美国赛默飞世尔科学公司)测定,在260/280条件下,使用NanoDrop ND-1000分光光度计(美国LabTech公司)测定吸光度为1.802。
对于RNA-seq,使用TRIzol试剂(Invitrogen, MA, USA)按照制造商的方案提取总RNA。RNA完整性使用Agilent 2100生物分析仪(Agilent Technologies, CA, USA)进行评估。我们研究中使用的样本的RNA完整性数(RIN)大于8.5。
我们生成了89.62 Gb的Hi-C raw reads,有效率为99.26%。Hi-C reads的Q20和Q30基础质量分别为96.74%和91.82%。
代码的可用性
本研究中没有使用特定的代码。数据分析使用方法中指定的标准生物信息学工具。
参考文献
杨,H。等.三种黄花鱼(Perciformes, Sciaenidae)全线粒体基因组序列的特征和对系统发育的新见解。Int。理学。19, 1741(2018)。
邹,W。等.饲料中添加维生素C对朱花鱼幼鱼生长性能、体成分及生化参数的影响(Nibea coibor).Aquac。减轻。26, 60-73(2020)。
黄玉生,温晓斌,李世凯,宣学忠,朱德生。蛋白质水平对楚氏黄花鱼幼鱼生长、饲料利用、体成分、氨基酸组成及生理指标的影响。Nibea coibor.Aquac。减轻。23, 594-602(2017)。
李,Z。等.益生元混合物对楚氏黄花鱼幼鱼生长性能、肠道菌群及免疫反应的影响Nibea coibor.鱼贝类免疫。89, 564-573(2019)。
黄燕,文欣,李生,李伟,朱东。饲料脂肪水平对楚氏黄花鱼幼鱼生长、饲料利用、体成分、脂肪酸谱和抗氧化参数的影响Nibea coibor.Aquac。Int。24, 1229-1245(2016)。
荣,H。等.羟脯氨酸对朱氏黄花鱼幼鱼生长性能、体成分、氨基酸谱、血液生化和胶原蛋白合成的影响(Nibea coibor).Aquac。Res。51, 1264-1275(2020)。
黄,Y.-S。等.共轭亚油酸对楚花鱼幼鱼生长、体成分、抗氧化状态、脂质代谢及免疫参数的影响Nibea coibor.Aquac。Res。49, 546-556(2018)。
黄燕,温霞,李生,李伟,朱东。棕榈油替代鱼油对朱氏黄花鱼幼鱼生长、饲料利用率、生化成分和抗氧化状态的影响。Nibea coibor.J.世界水族。Soc。47, 786-797(2016)。
林,F。等.饲料硒对楚花鱼幼鱼生长性能、抗氧化状态及组织硒沉积的影响(Nibea coibor).水产养殖536, 736439(2021)。
黄,Y。等.楚氏黄花鱼Δ6脂肪酸酰基去饱和酶的克隆、组织分布、功能表征及营养调控Nibea coibor.水产养殖479, 208-216(2017)。
林,Z。等.黄花鱼脂肪酰基Elovl5延长酶的克隆、组织分布、功能表征及营养调节Nibea coibor.基因659, 11-21(2018)。
张东,邵勇,姜松,李娟,徐晓霞。Nibea coibor生长激素基因的系统发育意义、微卫星变异及表达分析。内分泌素。163, 233-241(2009)。
单斌,赵丽娟,高涛,陆海华,闫艳Nibea coibor(鲈形目:石首鱼科)。线粒体DNA Part A27, 1681-1682(2016)。
Korlach, J. & Turner, S. W.单分子测序。在生物物理百科全书(罗伯茨编)2344-2347(施普林格,2013)。
道,W。等.两种罗非鱼的高质量染色体水平基因组揭示了它们的重复序列和性染色体的进化。摩尔。生态。Resour。21, 543-560(2021)。
朱,K。等.黄鳍鲷染色体水平的基因组组装(Acanthopagrus latus;Hottuyn, 1782)提供了关于其渗透调节和性别逆转的见解。基因组学113, 1617-1627(2021)。
黄,Y。等.斑点猫的染色体水平基因组组装(Scatophagus argus).基因组医学杂志。另一个星球。13, evab092(2021)。
周勇,秦伟,钟宏,张慧,周丽,张慧。鲤形目:鲤科(Hypophthalmichthys molitrix)基因组的染色体水平组装为其生态适应提供了新的视角。基因组学113, 2944-2952(2021)。
温格,上午。等.精确的循环一致性长读测序提高了人类基因组的变异检测和组装。生物科技Nat。》。37, 1155-1162(2019)。
Nurk, S。等.HiCanu:从高保真长读得到的片段复制、卫星和等位基因变体的精确装配。基因组Res。30., gr.263566.120(2020)。
下巴,c。等.分阶段二倍体基因组组装单分子实时测序。Nat方法。13, 1050-1054(2016)。
程浩,李国涛,冯晓霞,张海华,李海华。基于相位装配图的单倍型解析从头装配。Nat方法。18, 170-175(2021)。
Pueschel, R., Coraggio, F. & Meister, P.从单基因到全基因组:寻找核组织的功能。发展143, 910(2016)。
拉巴尔,f.a.。等.突破HiFi组合的极限揭示了两者之间着丝粒的多样性拟南芥基因组。预印在https://doi.org/10.1101/2022.02.15.480579(2022)。
Martin, M. Cutadapt从高通量测序读取中移除适配器序列。EMBnet.journal17, 10(2011)。
贝尔顿,j.m.。等.Hi-C:一种捕捉基因组构象的综合技术。方法58, 268-276(2012)。
北卡罗来纳州杜兰德。等.Juicer提供了一键式系统,用于分析循环分辨率Hi-C实验。细胞系统。3., 95-98(2016)。
Dudchenko, O。等.使用Hi-C从头组装埃及伊蚊基因组产生染色体长度支架。科学356, 92-95(2017)。
史蒂文,W。等.HiCUP:映射和处理Hi-C数据的管道。F1000res4, 1310(2015)。
Durbin, L. R.快速和准确的短读对齐与Burrows-Wheeler变换。生物信息学25, 1754-1760(2009)。
北卡罗来纳州杜兰德。等.Juicebox为Hi-C接触地图提供了无限缩放的可视化系统。细胞系统。3., 99-101(2016)。
梅恩,李志刚,李志刚,李志刚。人类端粒序列(TTAGGG)n在脊椎动物中的保护作用。Proc。国家的。学会科学。美国86, 7049-7053(1989)。
Marçais, G. & Kingsford, C.一种快速,无锁的k-mers发生的有效并行计数方法。Bioinforma。Oxf,心血管病。27, 764-770(2011)。
Manni, M., Berkeley, M. R., Mathieu, S., Simo, F. A. & Zdobnov, E. M. BUSCO更新:新颖和精简的工作流程以及更广泛和更深入的系统发育覆盖,用于真核生物、原核生物和病毒基因组的评分。摩尔。杂志。另一个星球。38, 4647-4654(2021)。
rihie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. merquury:基因组组装的无参考质量、完整性和分期评估。基因组医学杂志。21, 245(2020)。
或者,S。等.对可转置元素注释方法进行基准测试,以创建精简、全面的管道。基因组医学杂志.20.(2019)。
Flynn, J. M, Hubley, R., Goubert, C., Rosen, J. & Smit, A. RepeatModeler2用于转座因子家族的自动化基因组发现。Proc。国家的。学会科学。美国117, 9451-9457(2020)。
Grabherr, m.g.。等.三位一体:从RNA-Seq数据中重建一个没有基因组的全长转录组。生物科技Nat。》。29, 644-652(2011)。
哈斯,b.j.。等.利用最大转录本比对集改进拟南芥基因组注释。核酸测定。31, 5654-5666(2003)。
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1:使用genmark - et和AUGUSTUS进行无监督rna测序基因组注释。Bioinforma。Oxf,心血管病。32, 767-769(2016)。
使用基本本地对齐搜索工具(BLAST)安装。冷泉港。Protoc。2007, pdb。top17(2007)。
斯莱特,G. &伯尼,E.生物序列比较的启发式自动生成。BMC生物信息学6(2005)。
哈斯,b.j.。等.使用EVidenceModeler和程序组装拼接对齐自动真核基因结构注释。基因组医学杂志。9, r7(2008)。
卡马乔,C。等.BLAST+:架构和应用程序。BMC生物信息学10, 421(2009)。
汉族,Z。等.接近完整的基因组组装和黄鼓的注释(Nibea albiflora)可深入了解该物种的种群和进化特征。生态。另一个星球。9, 568-575(2019)。
Cai, M。等.具有多性染色体系统的鲤科鱼的染色体组装。科学。数据6, 132(2019)。
Emms, D. M. & Kelly, S. OrthoFinder:比较基因组学的系统发育矫形学推断。基因组医学杂志.20.(2019)。
Robert, C. & Edgar MUSCLE:具有高精度和高通量的多序列比对。核酸测定。32, 1792-1797(2004)。
从多个序列中选择保守块用于系统发育分析。摩尔。杂志。另一个星球。17, 540-552(2000)。
Gerard, T. & Jose, C.从蛋白质序列校准中去除发散和模糊对齐块后的系统发育改善。系统。医学杂志。56, 564-577(2007)。
沈玮,乐胜,李永和胡峰。SeqKit:一个跨平台的FASTA/Q文件操作工具包。《公共科学图书馆•综合》11, e0163962(2016)。
Koichiro, T., Glen, S. & Sudhir, K. MEGA11:分子进化遗传学分析版本11。摩尔。杂志。另一个星球。38, 3022-3027(2021)。
Sudhir, K., Glen, S., Michael, S. & Blair, H. S.时间树:时间线、时间树和发散时间的资源。摩尔。杂志。另一个星球。34, 1812-1819(2017)。
Letunic, I. & Bork, P.交互式生命树(iTOL) v5:系统发育树显示和注释的在线工具。核酸测定。49, 293-296(2021)。
Bernt, M。等.MITOS:改进的后生动物线粒体基因组注释。摩尔。Phylogenet。另一个星球。69, 313-319(2013)。
西弗斯,F。等.使用Clustal Omega快速,可扩展生成高质量的蛋白质多序列比对。摩尔。系统。医学杂志。7, 539(2011)。
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE:一种快速有效的估计最大似然系统发育的随机算法。摩尔。杂志。另一个星球。32, 268-274(2015)。
黄,D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. ufboo2:改进超快bootstrap近似。摩尔。杂志。另一个星球。35, 518-522(2018)。
序列读存档https://identifiers.org/insdc.sra:SRR19088065(2022)。
序列读存档https://identifiers.org/insdc.sra:SRR19088064(2022)。
序列读存档https://identifiers.org/insdc.sra:SRR19088063(2022)。
序列读存档https://identifiers.org/insdc.sra:SRR19088062(2022)。
叶克fenhazi, D. & Li, W.;基因库https://identifiers.org/insdc.gca:GCA_023373845.1(2022)。
李,W. & Yekefenhazi, D. nc_genemodel .gff3。figsharehttps://doi.org/10.6084/m9.figshare.19609608.v2(2022)。
确认
国家自然科学基金(no . 31872562)资助;福建省自然科学基金项目(2021J01829);国家重点研发计划项目(批准号2018YFD0900202)。
作者信息
作者及隶属关系
贡献
W.L.构想了这个计划。d.y., q.h., W.H.收集样本并提取基因组DNA和RNA。D.Y.和W.L.进行了数据分析并撰写了手稿。C.S.对数据分析做出了贡献。X.W.修改了手稿。所有作者都阅读并批准了手稿的最终版本。
相应的作者
道德声明
相互竞争的利益
作者声明没有利益竞争。
额外的信息
出版商的注意施普林格自然对出版的地图和机构从属关系中的管辖权主张保持中立。
补充信息
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议(Creative Commons Attribution 4.0 International License),允许以任何媒介或格式使用、分享、改编、分发和复制,只要您对原作者和来源给予适当的署名,提供知识共享许可协议的链接,并注明是否有更改。本文中的图像或其他第三方材料包含在文章的创作共用许可中,除非在材料的信用额度中另有说明。如果内容未包含在文章的创作共用许可协议中,并且您的预期使用不被法定法规所允许或超出了允许的使用范围,您将需要直接获得版权所有者的许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文

引用本文
Yekefenhazi, D., He, Q., Wang, X。et al。染色体水平的基因组组装Nibea coibor使用PacBio HiFi读取和Hi-C技术。科学数据9670(2022)。https://doi.org/10.1038/s41597-022-01804-6
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/s41597-022-01804-6
